
新闻中心

2023 年 12 月,中山大学肿瘤防治中心张力、杨大俊、赵洪云联合亚盛医药科研团队在《Cellular & Molecular Immunology》(IF=19.8,免疫学 TOP 期刊)发表题为《The BCL-2 inhibitor APG-2575 resets tumor-associated macrophages toward the M1 phenotype, promoting a favorable response to anti-PD-1 therapy via NLRP3 activation》的研究。PD‑1 单药抗肿瘤有效率有限,M2 型巨噬介导的冷肿瘤微环境是重要耐药原因;国产 BCL‑2 抑制剂 APG‑2575 的免疫协同机制此前未知。研究结合动物模型、细胞实验和肺癌临床样本开展探究,发现 APG‑2575 脱离 BCL‑2 靶点作用,直接结合 NF‑κB p65 激活 NLRP3 通路,促使 M2 巨噬向 M1 转换,通过 CCL5、CXCL10 招募 CD8⁺T 细胞,改善肿瘤微环境并增强 PD‑1 疗效,临床标本也印证相关标志物与免疫获益相关。该研究阐明 APG‑2575 联合免疫治疗新机制,为其肺癌联合用药临床试验提供理论支撑。
本研究利用细胞生长实时监测技术连续观测肿瘤细胞增殖活力,证实 APG-2575 在试验浓度下不能直接抑制肺癌细胞增殖、诱导肿瘤凋亡。该结果排除药物直接杀伤肿瘤的作用,从细胞层面佐证 APG-2575 的体内抑瘤效果源于重塑机体抗肿瘤免疫微环境。
PD-1 单药治疗非小细胞肺癌有效率仅 20%,M2 型肿瘤相关巨噬细胞造成的冷肿瘤微环境是免疫耐药关键,逆转巨噬极化可改善免疫疗效。
BCL-2 抑制剂 APG-2575 仅被证实可诱导血液瘤细胞凋亡,其协同 PD-1、调控巨噬的作用机制尚无研究。
本研究围绕 APG-2575 能否重编程巨噬、协同 PD-1,以及作用通路是否依赖 BCL-2 展开探究。
药理新发现:APG2575 可不依赖 BCL2 调控巨噬极化,突破该类药物仅靠促癌细胞凋亡的传统认知。
新作用靶点:首次证实 APG2575 直接结合 NFκB p65 并激活 NLRP3 通路,为同类药物研究提供新方向。
阐明完整通路:梳理出从 NFκB 激活到巨噬极化、趋化因子释放、招募 CD8⁺T、增敏 PD1 的完整作用链条。
临床落地:依托肺癌临床样本,确定多个可预判 PD1 药效的标志物,支撑联合用药临床试验开发。
研究从 PD-1 耐药与 APG-2575 联用机制未知的临床问题出发,先通过动物模型证实该药依托巨噬与 CD8⁺T 实现 PD-1 增效、无直接杀瘤作用;再经体外细胞试验明确药物调控巨噬极化、分泌趋化因子招募 T 细胞;继而分子实验探明其结合 NF-κB p65、激活 NLRP3 的非 BCL-2 依赖机制;最后借助肺癌临床样本验证标志物有效性,完成由表及里、基础结合临床的完整研究。
(一)体内药效结果
双动物模型证实协同抑瘤
APG 联合 PD-1 显著优于单药,肿瘤体积大幅下降;联合组肿瘤浸润活化 CD8⁺GZMB⁺/TNF-α⁺T 细胞显著增多,效应记忆 T(TEM、TRM)比例升高、初始 CD8⁺T 减少,肿瘤由冷变热。
APG 抗肿瘤完全依赖 CD8⁺T,
不依赖肿瘤细胞 BCL-2
在 CD8⁺T 细胞缺失或裸鼠体内,APG 单用及联合用药均失去抗肿瘤效果,清除 CD4⁺T 细胞则不影响药效;同时无论肿瘤细胞 BCL-2 过表达或敲除,APG 联合疗效都没有变化,TCGA 数据库也证实免疫治疗获益与非获益患者的肿瘤 BCL-2 表达无明显区别,且细胞实验证实 5μM 浓度 APG 无法诱导 LLC、H1299 癌细胞凋亡、不存在直接杀瘤作用,综上说明 APG 依靠调节机体免疫实现抗肿瘤效果。
巨噬清除 abolish 药效
PLX3397 清除巨噬后,APG 提升 CD8 浸润、协同 PD-1 的作用完全消失,APG 作用靶点是巨噬而非肿瘤 / T 细胞;体外 APG 直接孵育 CD8⁺T,T 增殖 / 活化无变化,排除直接作用 T 细胞。

图1. 在人源化 CD34⁺小鼠与 C57BL/6 小鼠模型中,APG-2575 可增强免疫检查点抑制剂的抗肿瘤疗效。

图2. APG-2575 的抗肿瘤作用依赖 CD8⁺T 细胞,该药通过调控巨噬细胞诱导 CD8⁺T 细胞发挥抗肿瘤免疫效应。
(二)巨噬极化与趋化因子结果
APG 不会改变巨噬细胞总数,仅调控巨噬极化,下调 M2 型标志物、上调 M1 型标志物,单细胞测序也印证其减少抑制型巨噬、增加促炎 M1 巨噬;同时 APG 促使 M1 巨噬高表达 CCL5 与 CXCL10 两种趋化因子以招募 CD8⁺T 细胞,在体内阻断这两种趋化因子后,药物促 T 细胞浸润及联合增效的作用便完全丧失。

图3. APG-2575 重塑巨噬细胞与 CD8⁺T 细胞的转录组特征,并通过趋化因子 CCL5、CXCL10 促进 CD8⁺T 细胞浸润。

图4. APG‑2575 可有效将 M2 型巨噬细胞重极化为 M1 表型。
(三)分子通路结果(核心机制)
APG 调控巨噬向 M1 极化的作用不依赖巨噬或肿瘤细胞的 BCL‑2,颠覆了该药仅靶向 BCL‑2 起效的传统认知;机制上 APG 直接结合 NF‑κB p65 关键氨基酸位点促使其入核,入核后的 p65 结合 NLRP3 启动子并上调其转录,进而激活 NLRP3 炎症小体、活化 Caspase‑1,促成 IL‑1β 等促炎因子释放,推动 M2 巨噬向 M1 转化并分泌 CCL5、CXCL10,最终招募大量 CD8⁺T 细胞;而使用 NLRP3 抑制剂 INF39 与 NF‑κB 抑制剂 JSH23 能够逆转上述巨噬极化与趋化因子上调,在动物体内还会削弱 APG 联合 PD‑1 的抗肿瘤效果。

图5. APG‑2575 通过上调 NLRP3 的表达促进巨噬细胞向 M1 型极化。

图 6. APG-2575 通过促进 NF-κB 入核,进而诱导 NLRP3 基因转录。
(四)临床样本结果
NSCLC 免疫获益患者:p-NFκB p65↑、NLRP3↑、CD86(M1)↑、CD206(M2)↓;高 NLRP3/p-p65/CD86 组 PFS、OS 显著优于低表达组,M2 高表达患者预后差;从临床层面佐证 NF-κB/NLRP3/M1 轴是免疫获益标志物。
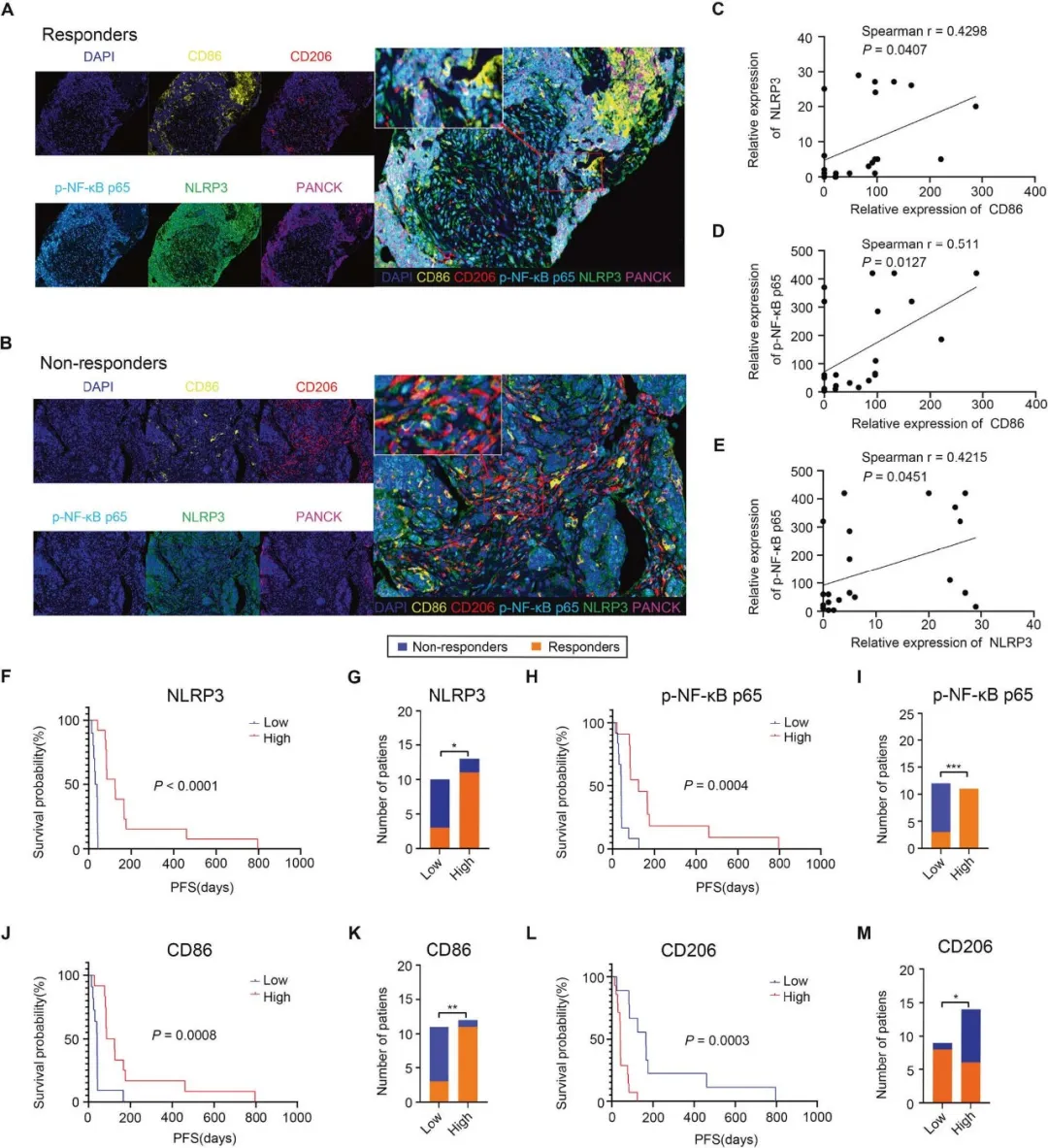
图 7. NSCLC 患者多色免疫荧光、生存预后分析数据。
1. 转化价值高:为 APG-2575 联合 PD-1 治疗肺癌的临床试验提供临床前支撑,助力破解 PD-1 耐药难题。
2. 实验设计严谨:依托动物、细胞、分子、临床样本多维度验证,多类前沿技术与正反对照试验保障结果可靠。
3. 理论创新:建立 BCL-2 抑制剂调控巨噬的全新联用思路,为各类同类药克服实体瘤免疫耐药提供新方向。
4. 研发新思路:发现药物有益脱靶作用,为新药研发发掘功能性脱靶靶点提供参考。
本研究采用细胞生长实时监测技术,动态检测肺癌细胞增殖活力,证实 APG2575 不能直接杀伤肿瘤细胞,排除药物直接抗肿瘤作用,佐证药物药效依靠免疫调控实现。

图8. 采用 细胞生长实时监测技术检测 APG‑2575 给药 72 小时期间的细胞增殖情况。
动态观测细胞状态:连续实时监测 LLC、H1299 肿瘤细胞增殖变化,直观观察给药后细胞生长趋势。
排除直接细胞毒性:证实 5μM 浓度 APG2575 无法抑制肿瘤增殖、诱导凋亡,明确无直接体外杀瘤活性。
支撑机制推论:从细胞层面证明 APG2575 体内抑瘤效果不源于直接靶向癌细胞,而是通过重塑肿瘤免疫微环境起效。
参考文献:
Luo F , Li H , Ma W ,et al.The BCL-2 inhibitor APG-2575 resets tumor-associated macrophages toward the M1 phenotype,promoting a favorable response to anti-PD-1 therapy via NLRP3 activation[J].Cellular & Molecular Immunology, 2024, 21(1):60-79.
